Prenatal choline and the development of schizophrenia
Abstract
Background
The primary prevention of illness at the population level, the ultimate aim of medicine, seems out of reach for schizophrenia. Schizophrenia has a strong genetic component, and its pathogenesis begins long before the emergence of psychosis, as early as fetal brain development. Cholinergic neurotransmission at nicotinic receptors is a pathophysiological mechanism related to one aspect of this genetic risk. Choline activates these nicotinic receptors during fetal brain development. Dietary supplementation of maternal choline thus emerges as a possible intervention in pregnancy to alter the earliest developmental course of the illness.
Aim
Review available literature on the relationship of choline supplementation or choline levels during pregnancy and fetal brain development.
Methods
A Medline search was used to identify studies assessing effects of choline in human fetal development. Studies of other prenatal risk factors for schizophrenia and the role of cholinergic neurotransmission in its pathophysiology were also identified.
Results
Dietary requirements for choline are high during pregnancy because of its several uses, including membrane biosynthesis, one-carbon metabolism, and cholinergic neurotransmission. Its ability to act directly at high concentrations as a nicotinic agonist is critical for normal brain circuit development. Dietary supplementation in the second and third trimesters with phosphatidyl-choline supports these functions and is associated generally with better fetal outcome. Improvement in inhibitory neuronal functions whose deficit is associated with schizophrenia and attention deficit disorder has been observed.
Conclusion
Prenatal dietary supplementation with phosphatidyl-choline and promotion of diets rich in choline-containing foods (meats, soybeans, and eggs) are possible interventions to promote fetal brain development and thereby decrease the risk of subsequent mental illnesses. The low risk and short (sixmonth) duration of the intervention makes it especially conducive to population-wide adoption. Similar findings with folate for the prevention of cleft palate led to recommendations for prenatal pharmacological supplementation and dietary improvement. However, definitive proof of the efficacy of prenatal choline supplementation will not be available for decades (because of the 20-year lag until the onset of schizophrenia), so public health officials need to decide whether or not promoting choline supplementation is justified based on the limited information available.
1. Introduction
Difficulties treating schizophrenia are found in all countries. Prenatal maternal starvation and infection are both well-established environmental risk factors for schizophrenia and call attention to this period as the earliest one in which a significant part of the risk for later schizophrenia occurs.[1,2] The foresight of several groups to collect sera from pregnant women has made it possible to conduct epidemiological studies of risk factors for schizophrenia in their offspring many years later. However, other than assuring general maternal nutrition and encouraging immunization, these epidemiological studies have not, as yet, resulted in recommendations for specific prenatal preventive interventions. This situation is not surprising as the greatest single risk factor for schizophrenia is genetic, accounting for over 50% of the risk.[3] The central role of genetic risk also points to the prenatal period, as many genes that convey risk for schizophrenia are expressed at much higher levels in the fetal brain than later in life.
[4] Infants do not express the symptoms of psychosis, so we must investigate the early development of abnormal brain function that might later become manifest as schizophrenia. Infants who manifested difficulties with limb movement in films taken by their parents during the first year of life – interpreted as evidence of a brain abnormality that occurred before birth – have an increased probability of subsequently developing schizophrenia.[5] Additional risk factors occurring during childhood, such as abuse and migration into a stressful urban environment, also appear to increase the risk for schizophrenia; however, no factor in childhood has been identified as a protective factor that reduces the risk of subsequent schizophrenia.[6,7] Some of the elevated gene expression in fetuses switches to adult levels soon after birth; this suggests that the brief window of prenatal development may be biologically irreversible. Thus, intervention in the prenatal period could potentially prevent developmental pathology that might later result in schizophrenia or other serious mental illness. However, primary prevention during the prenatal period necessarily means offering a potential preventive treatment for schizophrenia to over 100 pregnant women to prevent schizophrenia in the one person who would be expected to subsequently develop schizophrenia. The strategy of any preventive treatment is to affect early development of the pathophysiological basis of illness, but intervening during this critical prenatal window also means that unintended effects of the preventive treatment are impacting the most delicate period of development in the human life cycle. The possibility that results will not be known fully until decades later compounds the issue.
Nonetheless, models for the highly successful treatment of prenatal developmental defects exist. The most notable is the use of folate to reduce the incidence of cleft palate and other developmental issues related to the closing of midline structures, including spina bifida. Like mental illnesses, there are a number of genetic factors that influence this developmental problem. Testing for the genetic mutations would involve deep sequencing of many genes, with the possibility that some mutations might not be detected. Moreover, the mutation detection approach to prevention leaves parents with abortion of the fetus as the only possible remedy. Based on the findings of basic science studies showing the effects of folate on midline closure, maternal folate supplements became standard prenatal care for all women, regardless of the level of risk for cleft palate, spinal bifida, or other similar developmental abnormalities. It is notable that neither attention to diet alone nor supplements alone are as effective as the combination of both interventions.[8,9] The end result is that a difficult-to-treat developmental abnormality has seen a dramatic drop in incidence.
This review summarizes the emerging evidence that prenatal supplementation with choline might have a similar protective effect for schizophrenia and related mental disorders. This evidence might be relevant for public health initiative in countries, including China, that are concerned with the population-wide burden of mental illness. Because this possibility is just beginning to receive research attention as a target for intervention, there is little evidence to review systematically. A substantial portion of the interventional research has been conducted in only a few centers, including ours in Denver. Currently, neither prenatal choline nor any other specific prenatal intervention (except for multivitamins and folate) is recommended for either standard or highrisk prenatal care.
2. Methods
Medline was searched for key words ‘pregnancy’ or ‘fetal development’, both terms limited to humans. The resulting papers were then searched for articles that also contained the key word ‘choline’. The papers in the final set of 191 papers were then examined for relevance to dietary supplementation of pregnant women with choline or to the impact of choline levels during pregnancy of fetal brain development. As shown in Figure 1 and Table 1, eight papers were identified from 2012 onwards. There were no earlier papers that fit the two criteria of either measuring choline levels or intervening with choline and measuring an outcome related to cognition or risk for clinical illness. A parallel search of two major Chinese-language databases (CNKI and WanFang Data) identified no phosphatidyl-choline trials and the Chinese clinical test registration center (http://www.chictr.org/cn/) had no relevant trials. There are some intervention studies in China using folate supplementation, but they assess prevention of birth defects, not cognitive functioning. In China regular folate supplementation is recommended for pregnant women but there are no clinical recommendations for using phosphatidyl-choline.
Table 1.
Bibliography of selected articles about clinical and biological effects of maternal choline supplementation or of maternal choline blood levels
| Cheatham, 2012 [10] |
This randomized trial found no significant effects on cognition from prenatal choline supplementation. However, the placebo-treated women had unusually high levels of betaine, a choline metabolite, which suggests that they had consumed high levels of choline in their diet. The trial was double blind, but for ethical reasons participating women were informed that the goal of the trial was to increase their choline levels and that a diet rich in meat and eggs can do this. |
| Villamor, 2012 [11] |
This observational study found that maternal self-reported high dietary intake of choline (a methyl-donor nutrient) does not predict better cognition in the offspring at 3 years of age (see Boeke 2013 below). |
| Wu, 2012[12] |
This observational study was the first to find that both choline and the metabolite betaine are associated with cognitive function in offspring. |
| Jiang, 2012 [13] |
The assessment in this study is biochemical, but the gene methylation considered is associated with lower stress-reactivity, a positive clinical outcome. |
| Yan, 2012[14] |
This biochemical assessment identified one of the first biomarkers of choline effect that has been used in observational and interventional studies. |
| Wu, 2013[15] |
This paper shows that even though fish is generally lower in choline than mammal meat, liver, or eggs, in sufficient quantity it contains adequate choline content. |
| Boeke, 2013[16] |
This study is a follow-up of the 2012 paper by Villamor[11] which now shows cognitive effects into middle childhood of maternal self-reported high dietary choline. |
| Ross, 2013[17] |
This randomized intervention, conducted in an impoverished urban environment, found positive effects on cognition of choline supplementation during pregnancy. |
A general review of issues related to dietary choline supplementation in pregnancy is already available,[18] and an earlier review of the role of choline in fetal development focused on the results of animal model research that formed the rationale for a human test of phosphatidyl-choline supplementation (described below[19] ). Neither of these reviews considers outcomes research. We also reviewed other relevant basic science and clinical investigations of cholinergic mechanisms in development and in psychosis, including documents from the Institute of Medicine of the National Academies of Science of the United States concerning recommendations for dietary choline in pregnancy.[20]
3. Results
3.1. Alpha-7 nicotinic receptors in schizophrenia
Identification of choline as a possible factor in the development of schizophrenia arose from investigation of the neurobiological basis of inhibitory dysfunction in schizophrenia. Patients with schizophrenia describe an uncontrollable flooding of sensory information, particularly when they are acutely psychotic. At other times, they may purposefully withdraw from sensory stimulation and isolate themselves, or they may form hallucinations and delusions, which often seem to arise from attempts to comprehend stimuli that most people ignore. For example, a patient who cannot shut out others’ conversations around him from his awareness may conclude that people are talking about him. From a neurobiological perspective, a deficit in cerebral inhibition is a possible underlying neuronal mechanism. One test of inhibition is to examine the decrement in response when two stimuli are presented in close succession.[21] The first stimulus excites a set of neurons, but it also activates inhibitory neurons whose activity can last for up to 10 seconds. Generally the most robust inhibition is within the first 500 ms as GABAergic inhibitory neurons are activated. The test of the magnitude of inhibition is then the response to the second stimulus, which elicits less excitatory response because of the inhibition activated by the first stimulus.
We and others have studied neuronal inhibition in schizophrenia using auditory stimuli and measuring neuronal response using auditory evoked response recorded electroencephalographically. P50, a positive wave occurring 50 ms post stimulus, is a useful marker. Most normal subjects diminish the response to the second of a pair of stimuli by at least 50%, whereas most patients with schizophrenia do not. Not only are patients in the family affected, but one parent and half the unaffected siblings are also affected.[22-24] Genomewide linkage identifies a locus at chromosome 15q13 in the CHRNA7 gene.[22] A combination of pharmacological and genetic studies in rodent models of human P50 auditory evoked responses identified CHRNA7, the gene that forms the alpha-7 nicotinic acetylcholine receptor subunits, as a candidate gene for the deficit in neuronal inhibition.[22] Subsequent molecular studies of patients with schizophrenia have identified several deficits in Caucasians, Asians, and Africans, including single nucleotide polymorphisms (SNPs) in the gene’s promoter, abnormalities in a nearby partial gene duplication which can produce a truncated peptide that interferes with the assembly of alpha-7 nicotinic receptors, and copy number variations (CNVs) in which CHRNA7 is deleted.[25-28] All this evidence points to a genetically-determined deficiency in alpha-7 nicotinic receptors as a genetic factor in the onset and development of schizophrenia. Postmortem studies identify receptor decreases in the hippocampus, cerebral cortex, and the thalamus.[29,30] Many patients with schizophrenia smoke heavily, which can activate the alpha-7 nicotinic receptor. Compared to other nicotinic receptors, including those genetically associated with smoking , the alpha-7 nicotinic receptors require nearly three times the concentration of nicotine for activation, though it is also then inactivated or desensitized more quickly than the other receptors.[31]
3.2. Role of alpha-7 nicotinic receptors in brain development
In the hippocampus and frontal cortex, the level of CHRNA7 and the alpha-7 nicotinic receptor is almost 10-fold greater during fetal development than during adult life.[4,32] The high level of expression in the fetus drops suddenly in newborns and stays at the same relatively low level throughout adulthood. The high level in the fetus is not confined to inhibitory neurons (where most expression occurs in adult life) but also occurs in developing excitatory pyramidal neurons, where it covers the cell body and dendrites (Figure 2). After birth pyramidal neurons no longer express alpha-7 nicotinic receptors on their cell bodies and dendrites. Alpha-7 nicotinic receptors allow calcium ions to enter the cells, as do NMDA-type glutamate receptors. This influx of calcium appears necessary to stimulate the development of the mature form of the chloride ion transporter KCC2 (neuronal potassium-chloride symporter) that allows the neurons to hyperpolarize to their adult resting potential.[33,34] The immature embryonic form is not capable of enough chloride transport to hyperpolarize the neurons, so GABA is initially excitatory in fetal neurons but becomes inhibitory in adult neurons. The major window for this transition in humans is the second and third trimester of fetal life. In persons with schizophrenia, this transition does not fully occur, which may account for some of their inhibitory deficits after birth.[35,36]
During fetal development (left panel), cerebral alpha-7 nicotinic receptors are found on both pyramidal cells and interneurons. Choline in the extracellular fluid, rather than synpatic release of acetylcholine from cholinergic synapses (which have not yet reached the cerebrum), activates these fetal receptors. Postnatally (right panel), when cholinergic innervation has developed, acetylcholine activates the receptors, which are then restricted to interneurons. The activation of alpha-7 nicotinic receptors is required for the conversion of GABA from excitatory in fetal life to inhibitory in adult life and for the conversion of excitatory glutamate neurotransmisson from slower NMDA-type receptors to faster AMPA/kainate-type receptors.
3.3. Choline’s effects on neonatal pathophysiology associated with schizophrenia
Alpha-7 nicotinic receptors in the cerebrum do not receive their cholinergic innervation until just before birth, when the cholinergic afferents from the midbrain reach the hippocampus, thalamus, and cerebral cortex.[37] Therefore, acetylcholine is not the primary activator of the receptors during this period. Alpha-7 nicotinic receptors can also be activated by choline itself, at millimolar concentrations, many orders of magnitude higher than the concentrations required for activation by acetylcholine.[38,39] Once cholinergic synapses form (late in pregnancy), choline itself is no longer a significant source of activation of alpha-7 nicotinic receptors, so choline levels do not impact cholinergic neurotransmission in adults. However, in fetal life the concentration of choline does affect the activation of alpha-7 nicotinic receptors.
The necessary millimolar concentrations of choline are normally found in the amniotic fluid. However, choline has many uses in fetal life, principally as a central component in the synthesis of cell membranes (which requires large quantities of phosphatidyl-choline), but also as substituent along with folate in the one-carbon metabolism cycle. Although choline deficiency does not occur in non-pregnant adult women, it is estimated that approximately 30% of women are choline deficient during pregnancy because of the extraordinary requirements for choline by the fetus.[18] About onethird the necessary choline can by synthesized by the woman herself; the remainder requires dietary intake. The principal regulatory enzyme is PEMT, phosphatidylethanolamine N-methyltransferase. Common SNPS in the PEMT gene are associated with inability to produce extra choline during periods of deficiency. In Asians, these polymorphisms have been associated with increased rates of schizophrenia.[40]
Famine, which affects many nutrients, is one cause of choline deficiency. Famines in both China and in Holland have been associated with increased risk for later schizophrenia in the offspring of women who were pregnant during the famines.[2,41,42] Although individual nutrients were not assessed in either famine, the Chinese famine appears to have involved deprivation of nutrient-containing foods, while some caloric intake was maintained.[41,42] The Dutch famine appears to have resulted in nearly complete deprivation of both nutrients and overall calories. Choline is found in significant amounts in animal cell membranes, so meat or eggs are the most concentrated sources. Among non-animal sources, soybeans have the highest levels. The recommendations from various sources for dietary intake of choline acknowledge that there are few scientific studies that justify the current recommendations.[20]
Serum choline levels in normal women who are not in famine areas vary from 8-12 microM/L.[43] The reasons for this variance are several. One possibility is the genetic variation in PEMT, a second is variance in dietary intake of choline-containing foods, and a third is maternal stress. During stress, the woman preferentially holds choline in her own liver, which lowers the levels observed in the serum that is available to the fetus. Stress can occur for different biopsychosocial reasons, including stressful economic or social circumstances surrounding the pregnancy or in the community, maternal mental illnesses like anxiety and depression, and physical illnesses such as infection. All these factors have also been associated increased risk of subsequent schizophrenia among the women’s children.[44]
Nicotine is not known to affect choline levels, but it does have a complex interaction with alpha-7 nicotinic receptors.[45] A single high dose of nicotine, which can occur after smoking an initial cigarette, binds to the receptor and opens the pore that admits ions. However, the receptor pore then closes with the nicotine still bound to the receptor, which leaves the pore blocked rendering the receptor incapable of further activation. This inactivated or desensitized state persists in the continued presence of nicotine. Most regular smokers have high levels of nicotine in their bodies on a chronic basis; in a pregnant smoker the maternal nicotine would reach the fetus deactivating its alpha-7 nicotinic receptors.
The effects of these various risk factors for poor neuronal development and later schizophrenia can be observed by conducting cerebral electrophysiology on infants. We recorded the P50 response from newborns and a variant of the technique was used to record P50 evoked potentials in adults. During active sleep, when infants are in a cerebral state similar to rapid eye movement sleep in adults, P50 evoked potentials can be elicited; the mean level of inhibition in the paired pulse stimulation paradigm among infants is similar to that observed in adults. However, inhibition is decreased in infants with a psychotic father or mother, and in infants with a mother who is depressed, anxious, or a smoker.[46] Newborn inhibition thus reflects the pathophysiological effect of in utero exposure to factors that increase the offspring’s later risk for schizophrenia.[47-49] The longterm consequences of these deficiencies in inhibition in newborns can be observed when the offspring are assessed at 40 months of age, by which point reliable parental observations of the child’s behavior can be obtained.[50] Parents of children with normal cerebral inhibition at birth are significantly less likely to report symptoms of attention deficit disorder in their children 40 months after birth than those whose children had reduced inhibition at birth. (Figure 3)
Relationship of cerebral inhibition assessed by auditory evoked P50 ratio during the first month of life with the number of attentiondeficit hyperactivity symptoms at 40 months of age (reprinted with permission from Hutchison et al.[50])Fifty children were included in the study. The Child Behavior Checklist, completed by the parents, was used to assess the number of ADHD symptoms at 40 months of age. Inhibition at one month was a significant predictor of the number of attention deficit-hyperactivity symptoms at 40 months (F1,46 = 5.40, p = 0.025).
3.4. Randomized controlled trials of dietary choline supplementation in pregnancy to decrease risk for later mental illness
The pathophysiology of alpha-7 nicotinic receptor activation and associated clinical findings raise the possibility that an intervention to improve the development of cerebral inhibition in fetal life might decrease the risk for later schizophrenia. Both genetically diminished numbers of alpha-7 nicotinic receptors and deficiencies in choline might contribute to abnormalities in the development of cerebral inhibition. At this point there is no known way to increase alpha-7 nicotinic receptors if they are genetically deficient. Such deficiencies can occur because of mutations in the CHRNA7, but they also can occur from deficiencies in the other genes such as neuregulins, which are required to assemble alpha-7 nicotinic receptors.[51]
Choline can be manipulated by altering dietary intake. We first tried the experiment in animals, using a mouse strain with a genetically deficient CHRNA7, resulting in about 50% of the normal number of alpha-7 nicotinic receptors. The strain’s mutation affects only the expression, not the normal structure of the receptor. Supplemental dietary choline was provided at four times usual dietary amounts from conception through weaning for half the animals. Upon weaning, all animals were fed diets with usual amounts of choline until adulthood. At adulthood, those mice whose mothers had been supplemented had significantly greater cerebral inhibition than those whose mothers had received the usual diets.[52] The experiment raised the possibility that perinatal choline might overcome the genetic deficiency in alpha-7 nicotinic receptors and produce mice offspring with a permanent enhancement in cerebral inhibition. To determine if the effect was due to specific interaction with alpha-7 nicotinic receptors, the experiment was repeated in mice of the same strain whose alpha-7 nicotinic receptors were entirely removed by a CHRNA7 null mutation. These animals failed to show the enhancement of cerebral inhibition after perinatal choline supplementation, which indicates that the choline needs to interact with alpha-7 nicotinic receptors.[53]
Based on these animal model experiments, we secured permission from United States Food and Drug Administration (U.S. FDA) to conduct a placebocontrolled trial of choline supplementation in pregnant women.[17] The women were selected to have normal pregnancies with no history of fetal defects or known genetic abnormalities such as Down’s syndrome. The woman’s mental illness was not a consideration; about half had lifetime histories of anxiety or depression; one was psychotic. Many of the women were generally highly stressed by the pregnancy because of their relatively low socioeconomic status. Women who smoked were excluded as were those who used drugs but those who stopped smoking or using drugs because of pregnancy were accepted into the study. All women provided written informed consent, and the project was approved by the Colorado Multi-Institutional Internal Review Board and the National Institute of Mental Health.
Choline as a dietary nutrient is on the U.S. FDA ‘Generally Recognized as Safe’ List, so it does not require approval as a food additive, but since we were proposing an effect on future disease, approval was required and granted. All women received repeated dietary advice to eat food high in choline by an obstetrical nurse during regular visits. They also were referred for treatment of anxiety or depression, asked to take prenatal vitamins including high dose folate, and to receive vaccination against influenza and regular obstetrical care. The nurse and the pregnant woman were blind to choline or placebo assignment. Choline was administered as 7 capsules, 4 in the morning and 3 at night, each containing 900 mg of phosphatidylcholine. The total daily choline supplement was 900 mg, approximately twice the recommended dietary intake. Although choline in food is absorbed from the small intestine without problem, a bolus of choline administered as a supplement would reach the large intestine, where intestinal bacterial produce trimethylurea. While it is not harmful, some women experience a fishy odor which is distasteful. Phosphatidyl-choline is well absorbed and the form most present in food. It is impervious to bacterial catabolism.
At birth, the infant was administered phosphatidylcholine drops, 700 mg per day, for an additional month if the mother had received choline during pregnancy; a placebo was administered to the infants whose mothers received placebo during pregnancy. Based on the results we obtained at 3 months showing that most infants, regardless of treatment, eventually develop some cerebral inhibition, in future trials we will not supplement the infants post birth. About half the women in both groups fed their babies with breast milk predominantly. There were no serious side effects that were observed in greater frequency in choline-treated pregnancies for either mother or baby. Babies regardless of treatment had a normal range of weight, head circumference, length, gestational age, and Apgar scores at birth and were normal for overall developmental assessment at 6 months.
Recordings of P50 evoked potentials were performed at one and three months after birth. Inhibition of the P50 potential in the paired stimulus paradigm by at least 50% was the pre-determined criterion for normal development of cerebral inhibition. Significantly more of the choline-treated newborns at one month achieved that criterion than the placebo-treated infants (Figure 4). At three months, there was no significant difference between the groups. The catch-up by the placebo-treated infants is typical of early developmental abnormalities, which, regardless of later significance, often disappear in early childhood. For example, movement abnormalities observed in films of infants who later developed schizophrenia also disappeared quickly. What is more concerning is that 20% of infants in both groups continued to have abnormal cerebral inhibition. These tended to be lower birth weight males, who are generally susceptible to an increase in developmental issues. We do not know if higher doses of choline would have increased their cerebral inhibition. It remains to be seen whether or not the choline-treated babies will have fewer attention deficit symptoms than the placebo-treated babies–as predicted from their one month P50 evoked potential. Of course, the long-term outcome in adulthood, including the risk for schizophrenia, will not be known for over two decades.
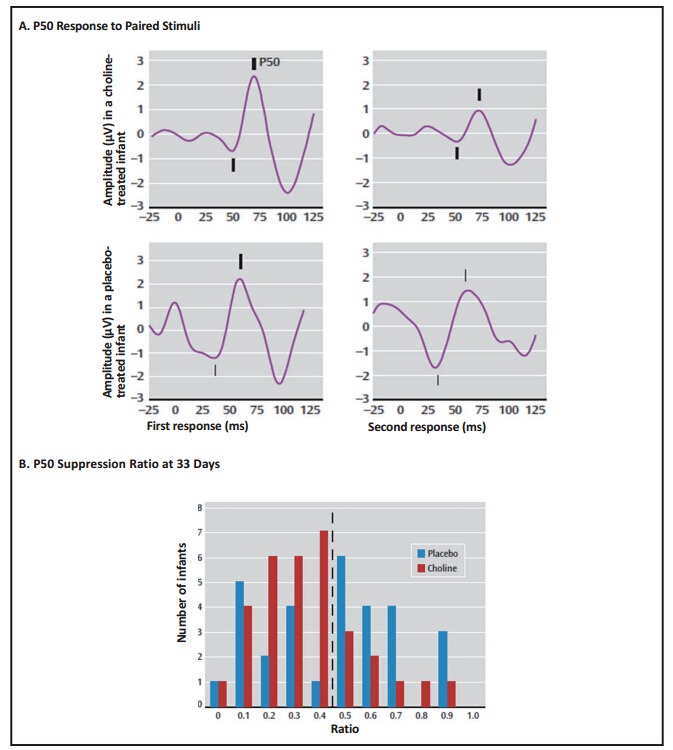
Panel B is a histogram of the P50 ratio at a mean adjusted age of 33 days. The dashed line demarcates the normal level of P50 inhibition, ratio 0.5. More choline than placebo-treated infants were in this normal range (χ2 = 6.90, df=1, p=0.009).
3.5. Other substances and other effects of choline in human prenatal development
Most studies of maternal conditions that affect the risk for schizophrenia rely on epidemiological methods, based on samples of maternal serum collected at or near birth 20 to 30 years earlier. None of these studies have examined choline content. Vitamin D levels show a biphasic relationship with higher levels and lower levels associated with increased risk for schizophrenia.[54] Omega-3 fatty acids and folate, though important nutrients, are not known to affect the fetal risk for schizophrenia, but have been shown to prevent the transition from prodrome to psychosis.[55]
Many women smoke early in the first trimester and then stop when they realize they are pregnant. We had ten women who smoked early in pregnancy but stopped when our nurse observed them. We excluded these women from the primary data analysis. Regardless of treatment, the infants generally had normal levels of cerebral inhibition at one month. A similar phenomenon has been observed with infant lung function, which also reflects nicotinic receptor activation. Infants whose mothers smoked early in pregnancy and then stopped have better pulmonary function because of their early nicotine exposure.[56] Once the nicotine itself, which desensitizes the receptors, is removed, the increased numbers of now active receptors appears to improve pulmonary development. At three months, however, these infants whose mothers smoked had abnormal cerebral inhibition, the only infants to lose inhibition once it had developed. We hypothesize that the loss of inhibition is due to second-hand smoke from the mother who likely resumed smoking sometime after giving birth.
Choline has other roles in fetal development, as noted before. Large amounts of choline are needed for the construction of cell membranes, but relatively low concentrations of choline are sufficient to achieve this.[57] At moderately higher levels, choline participates in one carbon metabolism; choline supplementation has been observed to increase DNA methylation, which would have possible effects on gene functions.[58] Choline’s role as an alpha-7 nicotinic agonist requires the highest concentrations, although no choline is consumed in the process. Evidence that it has a specific effect on choline receptors comes from genotyping the infants in our study. In the placebo-treated babies, a CHRNA7 polymorphism in the gene’s promoter that is associated with schizophrenia was also associated with decreased P50 inhibition, consistent with the hypothesis that the pathophysiological effect of the gene manifests early in development.[54] In the choline-treated babies, this effect was not observed; the infants had normal cerebral inhibition regardless of genotype, consistent with the hypothesis that dietary choline can overcome the genetic effect, as was previously seen in the animal model experiments.[52]
There has been one other randomized trial of prenatal choline to assess whether it had overall effects on cognitive function in infants.[10] A slightly lower dose of choline was administered, and the children’s cognition was tested at one year and not found to differ between the placebo and choline-treated groups. The results were significantly influenced by higher estimated betaine and choline in the diets of the placebo-treated women (betaine is intro-converted with choline). The women in this study had higher levels of education and socioeconomic status than the women in our study, which may have allowed them to eat better diets during their pregnancy. Assessment of maternal choline intake by dietary questionnaires in observational studies have not detected effects on childhood cognition at 3 years of age.[11] Thus, the more robust supplementation we used may be necessary to obtain significant effects. However, in a subsequent study of the same children at 7 years of age, estimated maternal choline intake above 400 mg/day did have significant effects on cognition.[16] Another observational study that measured free plasma choline and a related metabolite, betaine, in midsecond trimester found a range between 8.10 and 11.3 micromol/L, with a mean level of 9.40 micromol/L. The mean (sd) estimated choline dietary intake was 383 (99) mg/day, lower than the recommended 450 mg/day. In a multiple regression analysis of the results, both plasma free choline (Beta=6.05, SE=2.83, p=0.009) and betaine (Beta=7.35, SE=1.93, p=0.002) were significantly related to infant neurodevelopmental milestones at 18 months. Other nutrients, including vitamin B12 and folate, were not related.[12] An observational study that measured both serum free choline and phosphatidyl-choline[43] found high levels in relatively advantaged mothers; phosphatidyl-choline levels correlated highly with the child’s performance IQ at age 5, but the effect was not significant (r=0.77, p=0.28 for maternal levels at 18 weeks and r=0.92, p=0.25 for cord blood). Other studies of choline in pregnancy have focused on levels and effects on gene methylation. Higher dietary intake (930 mg/day versus 480 mg/ day) increased promoter methylation of the genes for corticotropin releasing factor and the glucocorticoid receptor.[13] Cord plasma cortisol levels were 53% lower for the infants whose mothers received the higher choline diets. In a second study, higher choline diets produced a doubling of the levels of the choline metabolite dimethylglycine in cord plasma, consistent with the participation of dietary choline in one-carbon metabolic pathways, including those that methylate DNA.[14] In a study of choline measurements during pregnancy in the context of impoverished diets conducted in Jamaica, estimated mean dietary choline intake was 278 (29) mg/day.[59] Mean plasma choline levels were 8.4 (0.4) micromol/L, which is the low end of the range observed in the United States.[59] A study with stable isotopes showed selective partitioning of choline in the fetus, for both cell membrane synthesis and for methylation pathways, confirming the tremendous fetal need for choline.[60]
4. Discussion
4.1. Main findings
The evidence base supporting the use of prenatal choline supplementation to prevent the subsequent development of schizophrenia and other mental illnesses is very limited, both because this is a new field that is just beginning to receive research attention and because of the 20-year lag between the intervention (prenatal choline supplementation) and the outcome of interest (schizophrenia). Several studies have identified the important role alpha-7 nicotinic receptors play in the development of neuronal inhibition and the need for high concentrations of choline during the second and third trimesters of pregnancy to activate these receptors. Animal studies and retrospective studies in humans suggest that prenatal choline supplementation and prenatal maternal levels of choline are associated with cognitive functioning in neonates and infants and with attention deficit symptoms in young children. Two randomized controlled trials of prenatal choline supplementation have had contradictory results: the first[10] found no significant differences in subsequent cognitive functioning and the second[17] (conducted by the author of this review) found significant improvement in measures of cerebral inhibition among infants in the choline-treated group.
4.2. Limitations
More trials are called for, but no trial can address the fundamental problem that the outcome of randomized intervention with choline compared to placebo will not have an observable preventive effect for schizophrenia until 20 to 30 years later, beyond the time frame of most experimental trials. A retrospective epidemiological study that examined the serum of women from 30 years ago, some of whom had births that resulted in an adult offspring with schizophrenia, would be helpful, but such a study has not been conducted.
Intermediate variables, such as attention deficit symptoms, that are observable before four years of age, are more promising because they could provide answers about the utility of prenatal choline supplementation much earlier. Although most children with such symptoms do not develop schizophrenia, most adults with schizophrenia develop attention deficit symptoms in childhood and many of them continue to have attention and cognitive difficulties prior to the onset of schizophrenia. Thus, the possible alleviation of attention deficit symptoms by prenatal choline supplementation could be viewed as favorable prognostic sign. Moreover, since attention deficit symptoms themselves are disabling, their absence in children whose mothers received prenatal choline supplementation would establish a benefit of choline that would support its use, regardless of any further preventative effect for adult illness.
4.3. Implications
The burden of schizophrenia in any country is enormous, in terms of cost of care, lost productivity and distress for the affected patient, and the possibility of violence towards the family and community. It may be several decades before it can be definitively proven whether or not prenatal choline supplementation affects the number of persons who ultimately develop schizophrenia. Before definitive proof, it may not seem economical to supplement choline pharmaceutically for all pregnant women, as we have done in our study. On the other hand, pharmaceutical folate supplementation, now routine, has substantially reduced expensive-totreat midline birth defects, so there is a potential risk of waiting to implement choline supplementation. The relatively short 6-month interval for choline supplementation and the lack of any significant side effects for this common nutrient suggest that intensive dietary intervention might be justifiable, even for the current generation of pregnant women, to prevent illnesses that have massive costs when they appear later. In communities where choline supplementation isn’t feasible, dietary recommendations to increase consumption of meat, liver, and eggs during the last 6 months of pregnancy would appear prudent. If meat and eggs are unavailable or not regularly eaten, encouraging the use of soy flour and the consumption of fish, which also contain significant amounts of choline (Table 2), may be sufficient to supplement choline for most pregnant women. [16]
Table 2.
Recommended choline intake during pregnancy and possible sources of choline
| Recommended daily intakea | ||
|---|---|---|
| Age of woman | Adequate Intake |
Upper Limit |
| Pregnant <18 years | 450 mg | 3000 mg |
| Pregnant >18 years | 450 mg | 3500 mg |
| Choline content of foodsb | ||
| Food | Serving size | Choline mg/serving |
| Beef liver | 1 slice | 420 |
| Egg | 1 egg | 120 |
| Beef | 100 gm | 90 |
| Chicken liver | 1 liver | 85 |
| Fish | 100 gm | 85 |
| Bacon or pork | 2 strips bacon | 70 |
| Chicken | 100 gm | 67 |
| Tofu | 120 ml (0.5 cup) | 36 |
| Cereal | 120 ml (0.5 cup) | 22 |
| Cauliflower | 1 floret | 5 |
| White rice | 120 ml (0.5 cup) | 1.5 |
| Recommended phosphatidyl-choline supplementsc | ||
| Supplement | Daily amount | Choline content |
| 900 mg capsule | seven capsules | 900 mg |
a recommendations of the Institute of Medicine of the National Academies of the United States[20]
b from the United States Department of Agriculture[61]
c recommendation for phosphatidylcholine supplementation from the most recent clinical study[54]
Biography
Robert Freedman, M.D., is a graduate of Harvard Medical School and trained at the National Institutes of Health and the University of Chicago. For the past 35 years he has been a faculty member at the University of Colorado Health Sciences Center, where he is Professor of Psychiatry and Pharmacology, Director of the Schizophrenia Research Center, and Chair of the Department of Psychiatry. Dr. Freedman has published over 300 scientific articles. He has received the A.E. Bennett Prize of the Society of Biological Psychiatry, the Edward Sachar Award of Columbia University, the William K. Warren Award of the International Congress of Schizophrenia Research, the Stanley Dean Award of the American College of Psychiatrists, the Merit Award of the National Institute of Mental Health, the American Psychiatric Association Research Award and its Distinguished Service Award, and the Distinguished Investigator Award of the National Association for Research in Schizophrenia and Affective Disorders. He is a member of the Institute of Medicine of the National Academies of Science and has served as Editor of the ‘American Journal of Psychiatry’ since 2006. He co-founded the Institute for Children’s Mental Disorders in 1999. Institute investigators under his direction have discovered genetic variants that affect the risks for serious mental disorders, including schizophrenia and bipolar disorder. Their investigations have led to new experimental treatments, currently in FDA-approved tests, for pregnant women and their newborn children to prevent abnormalities in early brain development that may lead to mental illness later in life.
Funding Statement
This work was funded by US NIMH Grant MHP50MH086383 and R01MH056539, and by gifts from the Jerome and Mary Rossick Kern and the Institute for Children’s Mental Disorders and from Cy and Lyndia Harvey and the Anschutz Family Foundation.
Registration: The randomized clinical trial of choline supplementation discussed in this review[17] was registered as Clinicaltrials. gov identifier: NCT00332124.
Conflict of Interest: Dr. Freedman has a patent on the genomic structure of CHRNA7 and SNPs in the promoter through the Department of Veterans Affairs. He derives no income from this patent.
Ethics approval: The randomized clinical trial of choline supplementation discussed in this review[17] was approved by the Colorado Multi-Institutional Internal Review Board and the National Institute of Mental Health.
Informed consent: All women who participated in the randomized clinical trial of choline supplementation discussed in this review[17] provided written informed consent to participate in the project.